SIMILAR EFFICACY TO LUCENTIS IN THE TREATMENT OF nAMD1,2
A Phase 3 Clinical Trial Comparing BYOOVIZ and Lucentis1,2


*
The endpoints were chosen in consultation with appropriate regulatory bodies. BCVA was the selected primary endpoint for FDA approval.1
†
Active CNV indicated presence of leakage and intra- or subretinal fluid and was confirmed by central reading center during screening.1
Primary Endpoint: Difference of LS Mean Change in BCVA Between BYOOVIZ and Lucentis at Week 81

Difference in LS mean change from baseline in BCVA at Week 8 in the full analysis set (FAS) (BYOOVIZ − Lucentis); whiskers represent the 90% CI that is contained within the predefined equivalence margins of −3 to 3 letters, represented by the dashed lines.
n=351 (BYOOVIZ), n=353 (Lucentis). The FAS included all randomized participants, excluding 1 inadvertently randomized participant who did not receive study drug.1
- At Week 8, visual acuity for BYOOVIZ was within the predefined equivalence margins compared with Lucentis1
- Adjusted treatment difference between groups was −0.8 letters (90% CI: −1.8 to 0.2 letters)1
Primary Endpoint: Difference of LS Mean Change in CST Between BYOOVIZ and Lucentis at Week 41

Difference in LS mean change from baseline in CST at Week 4 in per-protocol set (PPS) (BYOOVIZ – Lucentis); whiskers represent the 95% CI that is contained within the predefined equivalence margin of −36 to 36 μm, represented by the dashed lines.
n=342 (BYOOVIZ), n=338 (Lucentis).
- At Week 4, CST for BYOOVIZ was within the predefined equivalence margins compared with Lucentis1
- Adjusted treatment difference between groups was –8.4 μm (95% CI: –19.4 to 2.7)1
1-Year/ Study: Change From Baseline in BCVA at Each Time Point Through Week 52 in the FAS2*

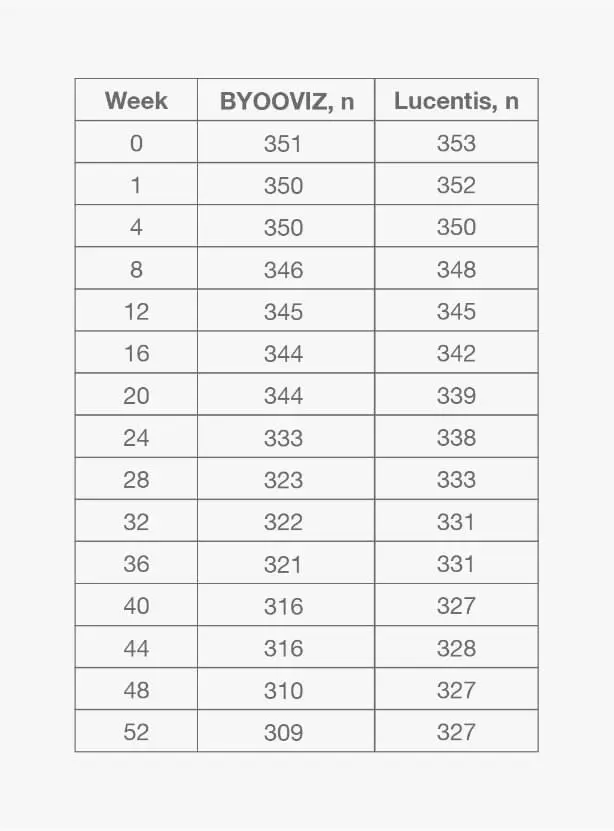
Mean (SD) change from baseline in BCVA for participants who completed Week 52 of the study: BYOOVIZ (n=309), 9.7 (11.4) letters; Lucentis (n=327), 10.4 (11.5) letters. Circles and squares represent mean and error bars represent SE at each time point.
*For additional information and all other secondary endpoints, please refer to Bressler NM, et al., 2021.2- Change from baseline in BCVA was 9.8 letters for BYOOVIZ and 10.4 letters for Lucentis at Week 52 (adjusted treatment difference [SE]: −0.6 [0.9]; 90% CI: −2.1 to +0.9 letters)2
- The proportion of participants who lost <15 letters and who gained >15 letters was comparable between treatment groups at all time points2
1-Year Study: Change From Baseline in CST at Each Time Point Through Week 52 in the FAS2

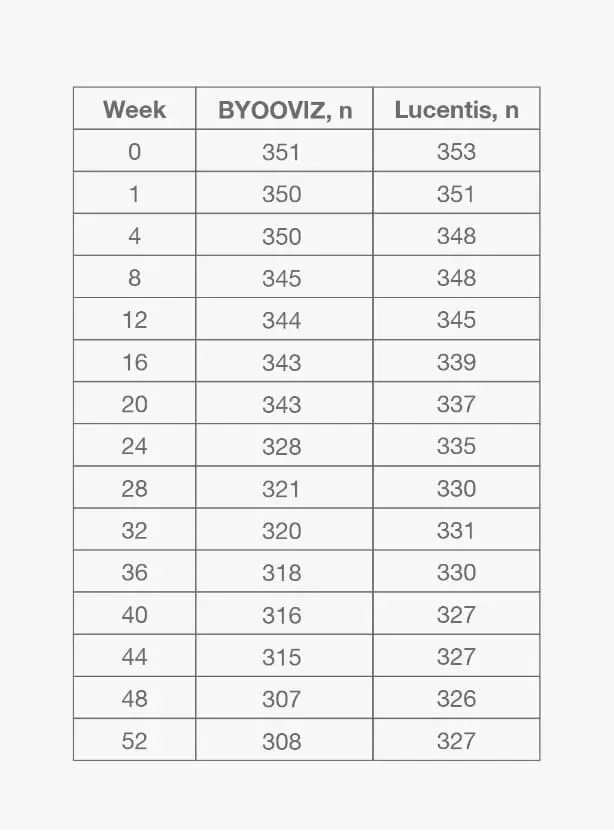
- Change from baseline in CST was −140.0 μm for BYOOVIZ and −125.1 μm for Lucentis at Week 52 (adjusted treatment difference [SE]: −14.9 [5.3]; 95% CI: –25.3 to –4.5)2
- The change from baseline in CST was comparable between treatment groups at all time points up to Week 522